Our collaborations focus on interdisciplinary work to elucidate the mechanisms behind diverse diseases. We have published several collaborative papers in high-impact journals such as Nature, Nature Medicine, and Cancer Discovery. These partnerships grant our lab access to several large cancer cohort studies with unique resources that are crucial for validating the novel statistical tools we develop.
Jian Hu, Kyle Coleman, Edward B. Lee, Humam Kadara, Linghua Wang, Mingyao Li
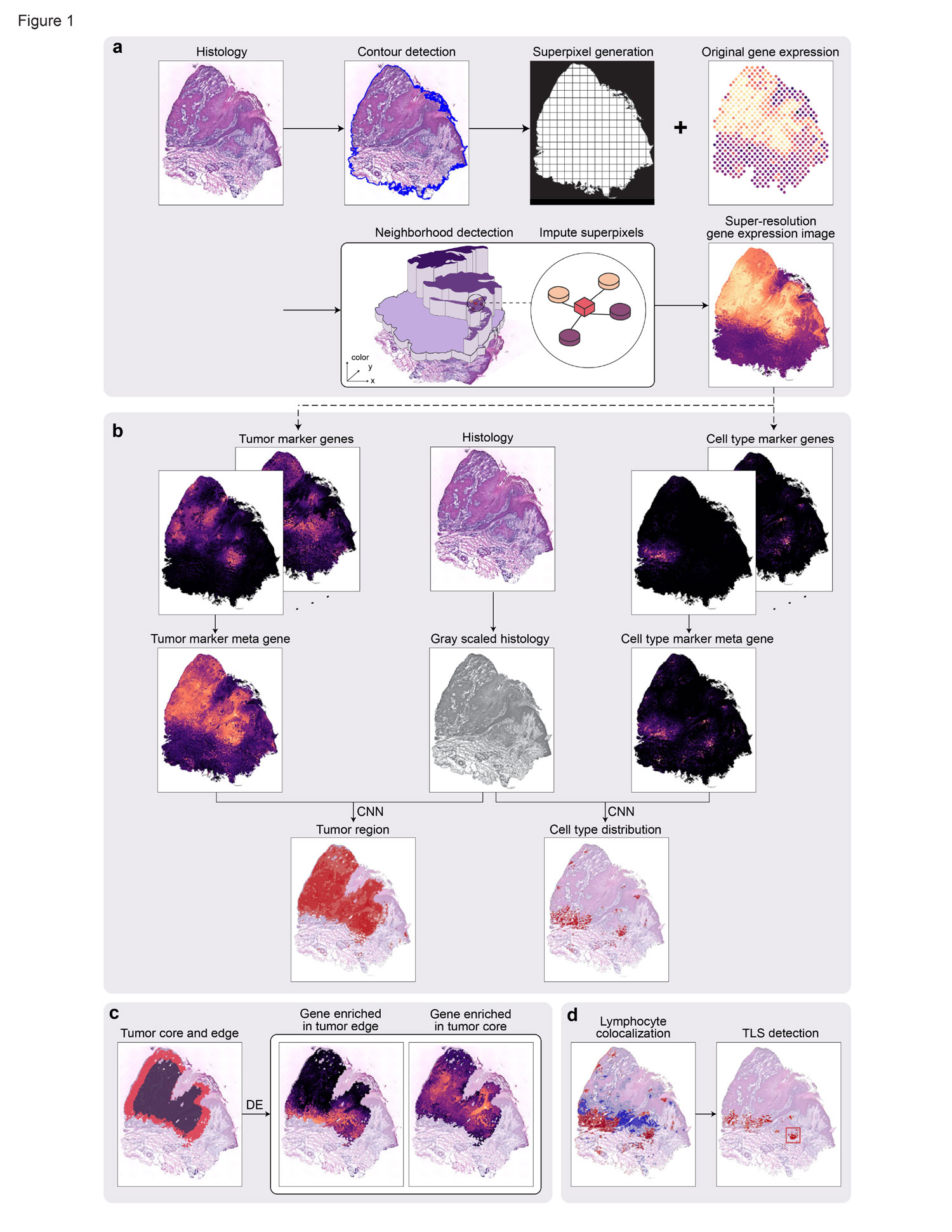
Spatial transcriptomics have enabled the comprehensive characterization of gene expression in tumor microenvironment. However, ST only measures expression in discrete spots, which limits their usefulness in studying the detailed structure of TME. Here we present TESLA, a machine learning framework for multi-level tissue annotation in ST. TESLA integrates histological information with gene expression to annotate heterogeneous immune and tumor cells directly on the histology image, and further detects tertiary lymphoid structures and differential transcriptome programs between the edge and core of a tumor. TESLA provides a powerful tool for understanding the spatial architecture of the TME.
Go to the article
Jian Hu, Xiangjie Li, Kyle Coleman, Amelia Schroeder, Nan Ma, David J Irwin, Edward B Lee, Russell T Shinohara, Mingyao Li
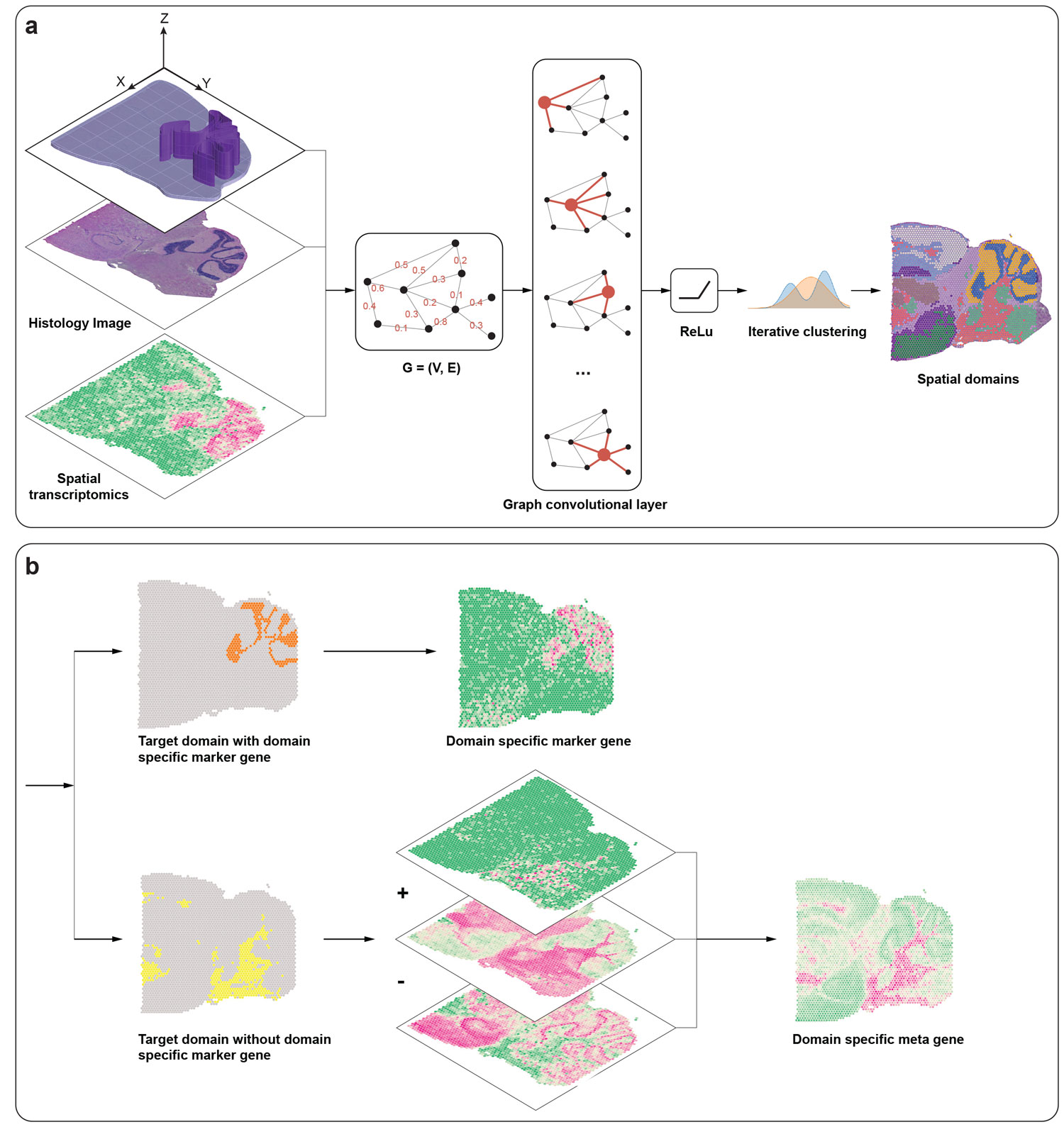
Recent advances in spatially resolved transcriptomics (SRT) technologies have enabled comprehensive characterization of gene expression patterns in the context of tissue microenvironment. To elucidate spatial gene expression variation, we present SpaGCN, a graph convolutional network approach that integrates gene expression, spatial location and histology in SRT data analysis. Through graph convolution, SpaGCN aggregates gene expression of each spot from its neighboring spots, which enables the identification of spatial domains with coherent expression and histology. The subsequent domain guided differential expression (DE) analysis then detects genes with enriched expression patterns in the identified domains. Analyzing seven SRT datasets using SpaGCN, we show it can detect genes with much more enriched spatial expression pat- terns than competing methods. Furthermore, genes detected by SpaGCN are transferrable and can be utilized to study spatial variation of gene expression in other datasets. SpaGCN is computationally fast, platform independent, making it a desirable tool for diverse SRT studies.
Go to the article
Jian Hu, Amelia Schroeder, Kyle Coleman, Chixiang Chen, Benjamin J Auerbach, Mingyao Li
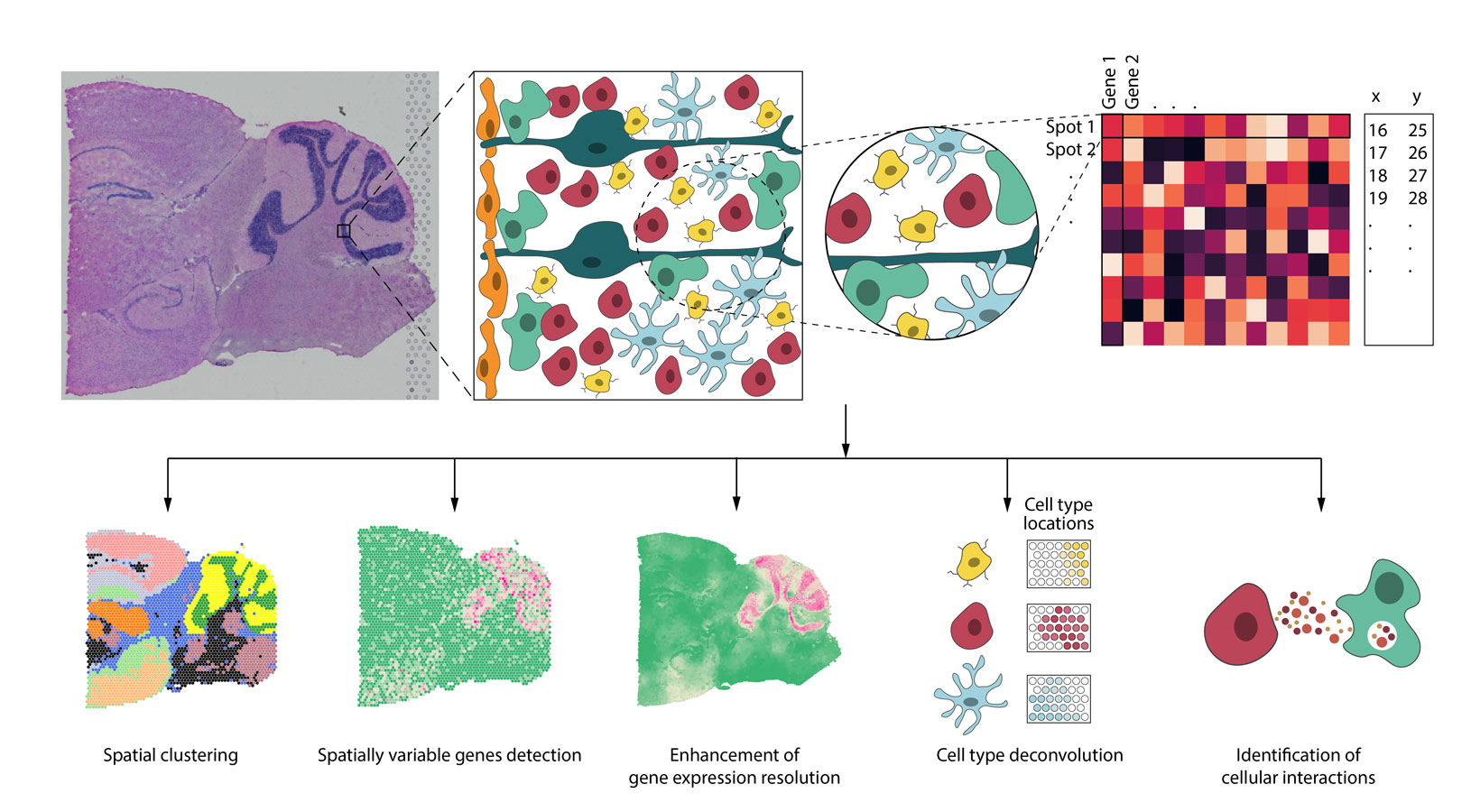
Spatial resolved transcriptomics (SRT) technologies have enabled scientists to get an integrated understanding of cells in their morphological context. Applications of these technologies in diverse tissues and diseases have transformed our views of transcriptional complexity. Most published studies utilized tools developed for single-cell RNA sequencing (scRNA-seq) for data analysis. However, SRT data exhibit different properties from scRNA-seq. To take full advantage of the added dimension on spatial location information in such data, new methods that are tailored for SRT are needed. Additionally, SRT data often have companion high-resolution histology information available. Incorporating histological features in gene expression analysis is an underexplored area. In this review, we will focus on the statistical and machine learning aspects for SRT data analysis and discuss how spatial location and histology information can be integrated with gene expression to advance our understanding of the transcriptional complexity. We also point out open problems and future research directions in this field.
Go to the article
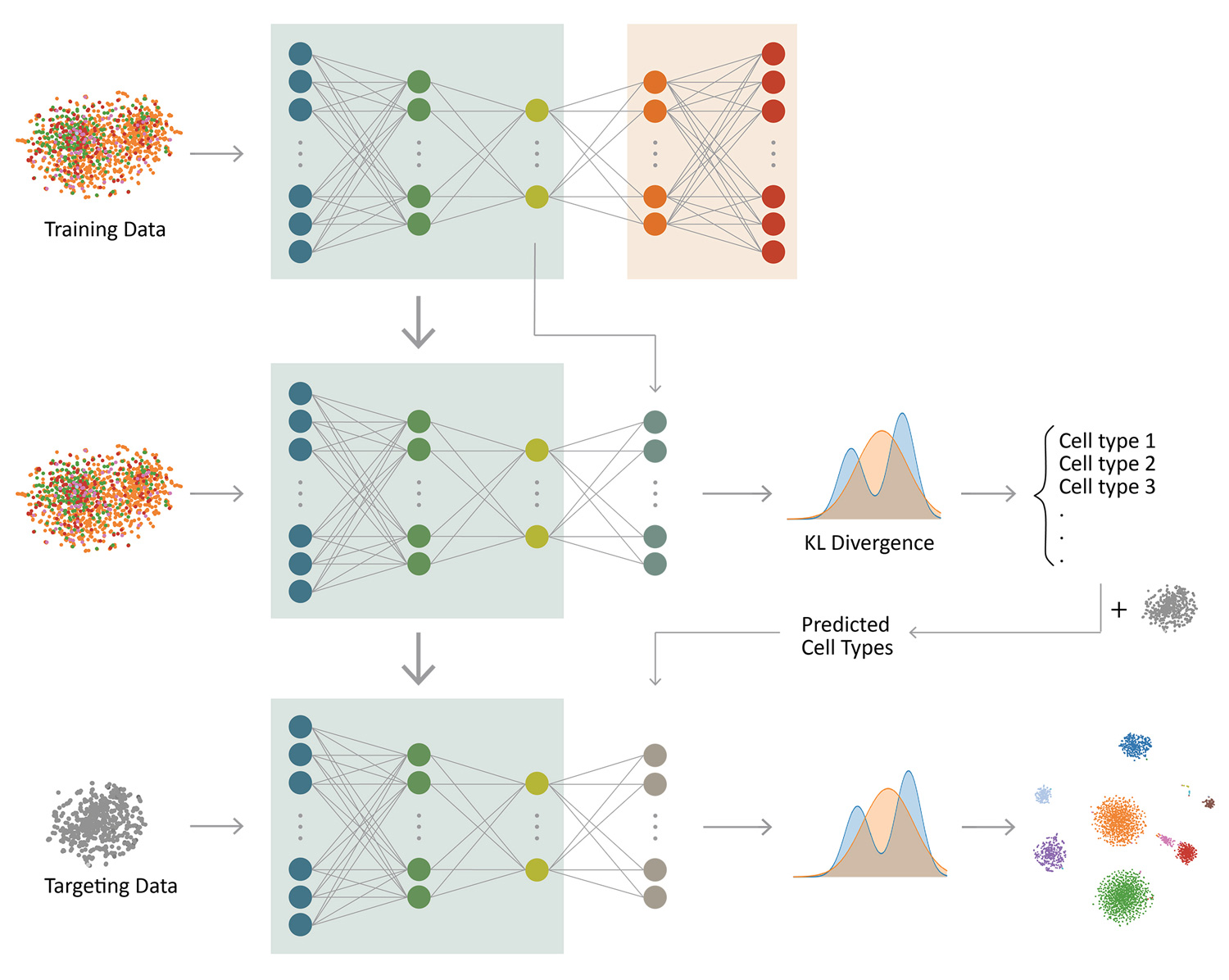
Jian Hu, Xiangjie Li, Gang Hu, Yafei Lyu, Katalin Susztak, Mingyao Li
Clustering and cell type classification are important steps in single-cell RNA-seq (scRNA-seq) analysis. As more and more scRNA-seq data are becoming available, supervised cell type classification methods that utilize external well-annotated source data start to gain popularity over unsupervised clustering algorithms; however, the performance of existing supervised meth- ods is highly dependent on source data quality and they often have limited accuracy to classify cell types that are missing in the source data. We developed ItClust to overcome these limitations, a transfer learning algorithm that borrows ideas from super- vised cell type classification algorithms, but also leverages information in target data to ensure sensitivity in classifying cells that are only present in the target data. Through extensive evaluations using data from different species and tissues generated with diverse scRNA-seq protocols, we show that ItClust considerably improves clustering and cell type classification accuracy over popular unsupervised clustering and supervised cell type classification algorithms.
Go to the article
Heng Xu, Zhu Zhu, Jian Hu, Jiawei Sun, Yan Wo, Xianshu Wang, Hongzhi Zou, Bin Li, Yixin Zhang
Keloids are an abnormal fibroproliferative wound-healing disease with a poorly understood pathogenesis, making it difficult to predict and prevent this disease in clinical settings. Identifying disease-specific signatures at the molecular and cellular levels in both the blood circulation and primary lesions is urgently needed to develop novel biomarkers for risk assessment and therapeutic targets for recurrence-free treatment. There is mounting evidence of immune cell dysregulation in keloid scarring. In this study, we aimed to profile keloid scar tissues and blood cells and found that downregulation of cytotoxic CD8+ T cells is a keloid signature in the peripheral blood and keloid lesions. Single-cell RNA sequencing revealed that the NKG2A/CD94 complex was specifically upregulated, which might contribute to the significant reduction in CTLs within the scar tissue boundary. In addition, the NKG2A/ CD94 complex was associated with high serum levels of soluble human leukocyte antigen-E (sHLA-E). We subsequently measured sHLA-E in our hospital-based study cohort, consisting of 104 keloid patients, 512 healthy donors, and 100 patients with an interfering disease. The sensitivity and specificity of sHLA-E were 83.69% (87/104) and 92.16% (564/612), respectively, and hypertrophic scars and other unrelated diseases exhibited minimal interference with the test results. Furthermore, intralesional therapy with triamcinolone combined with 5-fluorouracil drastically decreased the sHLA-E levels in keloid patients with better prognostic outcomes, while an incomplete reduction in the sHLA-E levels in patient serum was associated with higher recurrence. sHLA-E may effectively serve as a diagnostic marker for assessing the risk of keloid formation and a prognostic marker for the clinical outcomes of intralesional treatment.
Go to the article
Jian Hu, France W Fung, Marin Jacobwitz, Darshana S Parikh, Lisa Vala, Maureen Donnelly, Alexis A Topjian, Nicholas S Abend, Rui XiaoElectroencephalographic seizures (ES) occur in 10-40% of children with acute encephalopathy who undergo continuous EEG monitoring (CEEG), increasing evidence indicates that high ES exposure is associated with unfavorable neurobehavioral outcomes, and ES are often treatable with anti-seizure medications. However, given CEEG is resource-intense, a clinical prediction tool that enabled evidence-based targeting of CEEG resources to patients most likely to experience ES would be of great clinical value. In this study, we investigated on whether machine learning techniques would enhance our ability to incorporate key variables into a parsimonious model with optimized prediction performance for ES prediction in critically ill children. We analyzed data from a prospective observational cohort study of 719 consecutive critically ill children with encephalopathy who underwent clinically-indicated continuous EEG monitoring (CEEG). We implemented and compared three state-of-the-art machine learning methods for ES prediction: (1) random forest; (2) Least Absolute Shrinkage and Selection Operator (LASSO); and (3) Deep Learning Important FeaTures (Deep- LIFT). We developed a ranking algorithm based on the relative importance of each variable derived from the machine learning methods.Go to the article
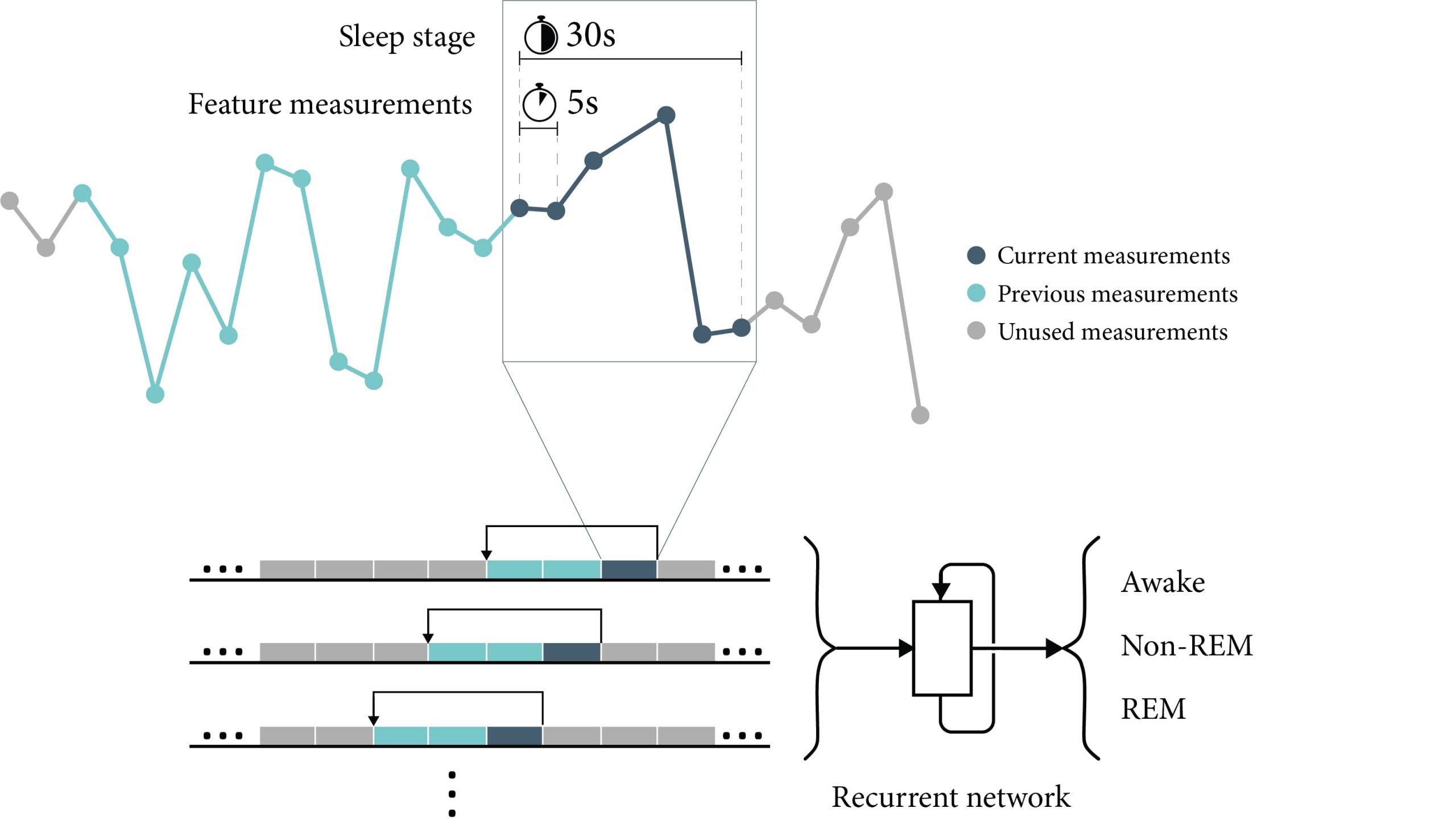
Jian Hu, Haochang Shou
The use of wearable sensor devices on daily basis to track real-time movements during wake and sleep has provided opportunities for automatic sleep quantification using such data. Existing algorithms for classifying sleep stages often require large training data and multiple input signals including heart rate and respiratory data. We aimed to examine the capability of classifying sleep stages using sensible features directly from accelerometers only with the aid of advanced recurrent neural network. In this study, we analyzed a publicly available dataset with accelerometry data in 5s epoch length and polysomnography assessments. We developed long short-term memory (LSTM) models that take the 3-axis accelerations, angles and temperatures from concurrent and historic observation windows to predict wake, REM and non-REM sleep.
Go to the article